<div id="toc"></div>
If anything is unclear: [sushi@fgcz.ethz.ch](mailto:sushi@fgcz.ethz.ch?subject=Question about SUSHI)
---
# How do I subset my DataSet to exclude/retain specific samples?
There are two possibilities:
- After starting an analysis you can use "samples" selection box to select/unselect the samples. This will not modify the parent DataSet but only the Child DataSet to be generated.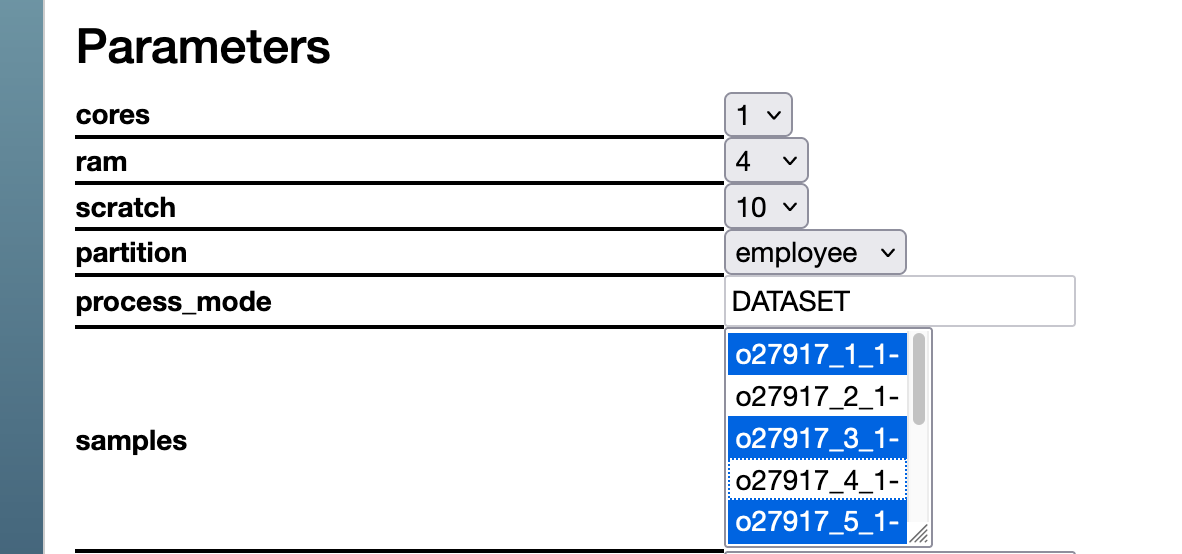
- On a dataset, clicking on "edit table" brings you to the "edit screen" that lets you remove samples from the DataSet using the minus signs on the left. Subsequently do "save as child" to generat a Child DataSet with the subset of samples. Clicking on "save" will permanently modify the current DataSet.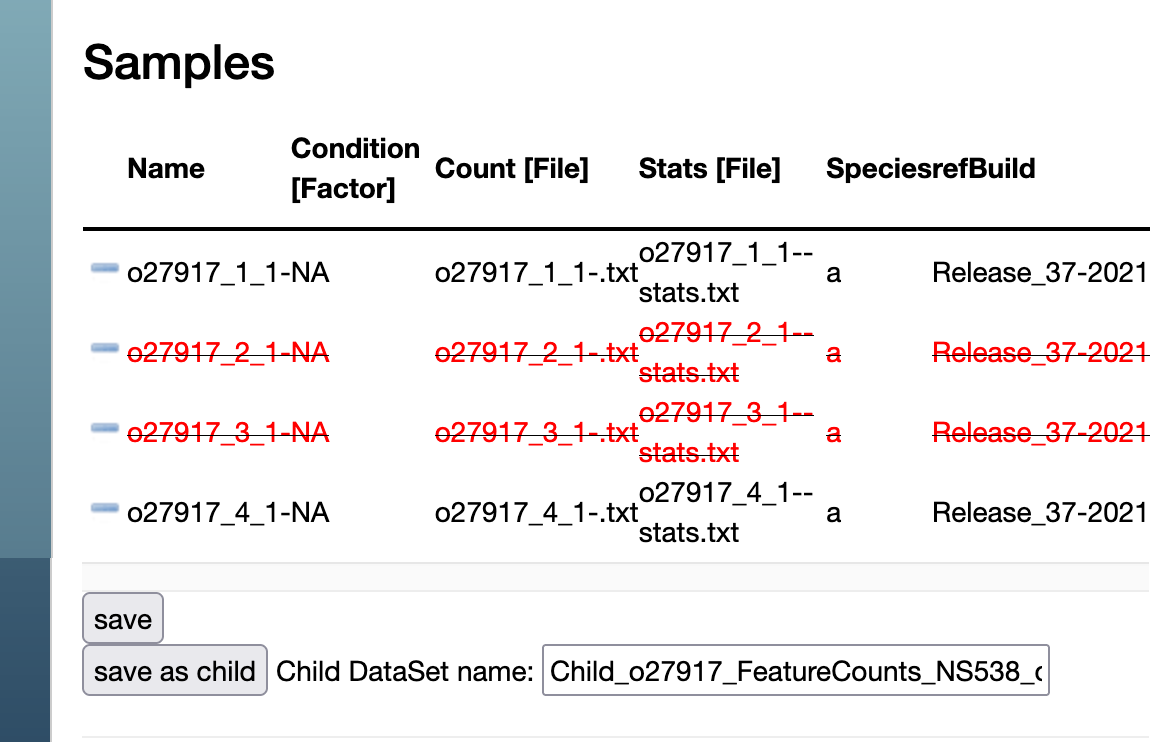
---
<br>
# How do I specify sample groupings for differential expression?
Groupings are specified by attributes (columns) in the dataset table. These columns must contain in the column name the tag "\[Factor\]".
# How do I add more columns/attributes of sample information to my DataSet, e.g., batch, sex?
- First, click the `edit table` to the right of the DataSet title.

- This will take you to the edit table page. Click the "+" at the very right of the table.
- You can also click the "edit table content" to modify any cell already in the table.
- You can also delete columns by clicking the "-" next to a column title.

- Once you've clicked to add a new column, the table will have a new empty column at the right.
- In the column title, give the column a name. **It must not contain spaces or any special characters *other* than "-" and "."**.
- **You must also ensure that you include " [Factor]" at the end of the column title!** *E.g.*, "Sex [Factor]".
- Then you can enter the data in the cells, again ensuring to avoid spaces and special characters.
- You can then save these changes to the current DataSet with the "Save" button, or save the changed table as a new child DataSet.
- You will then be returned to the main `edit table` page and can click the "Back to DataSet" at the top of the page.
- Any column labelled as `[Factor]` can be used in the differential expression tests.
---
<br>
# If I edit the DataSet (e.g., add a column, fix a spelling mistake), do my results change?
- No, if you edit the DataSet in any way, your child DataSets (and Static and Live Reports) remain unchanged, and you will need to run apps again to have the results reflect the modified DataSet.
---
<br><br>
# How do I know whether/which app parameters I can/should change?
- Critical parameters are marked as required in red.
- They may be autopopulated based on previous DataSet/B-Fabric, in this case, leave as default.
- For more routine analysis, *e.g.*, RNA seq, scRNA seq, ChIP seq, the defaults are mostly sufficient and can be considered 'best practice'.
- But if you have an idea of some specific parameters you need, don't be afraid to play around! If it's not clear where you need to put these parameters, just ask us.
- When trying different parameters, don't be alarmed if the app crashes, often we have these defaults for good reason!
- In cases where you've changed parameters, you should make good use of the "Comment" text box at the top of the app parameters page, to aid later identification of multiple versions of the same app but with different parameters
- If you are unsure of which parameters you need to use, *e.g.*, paired or strandMode for mapping, ask your coach!
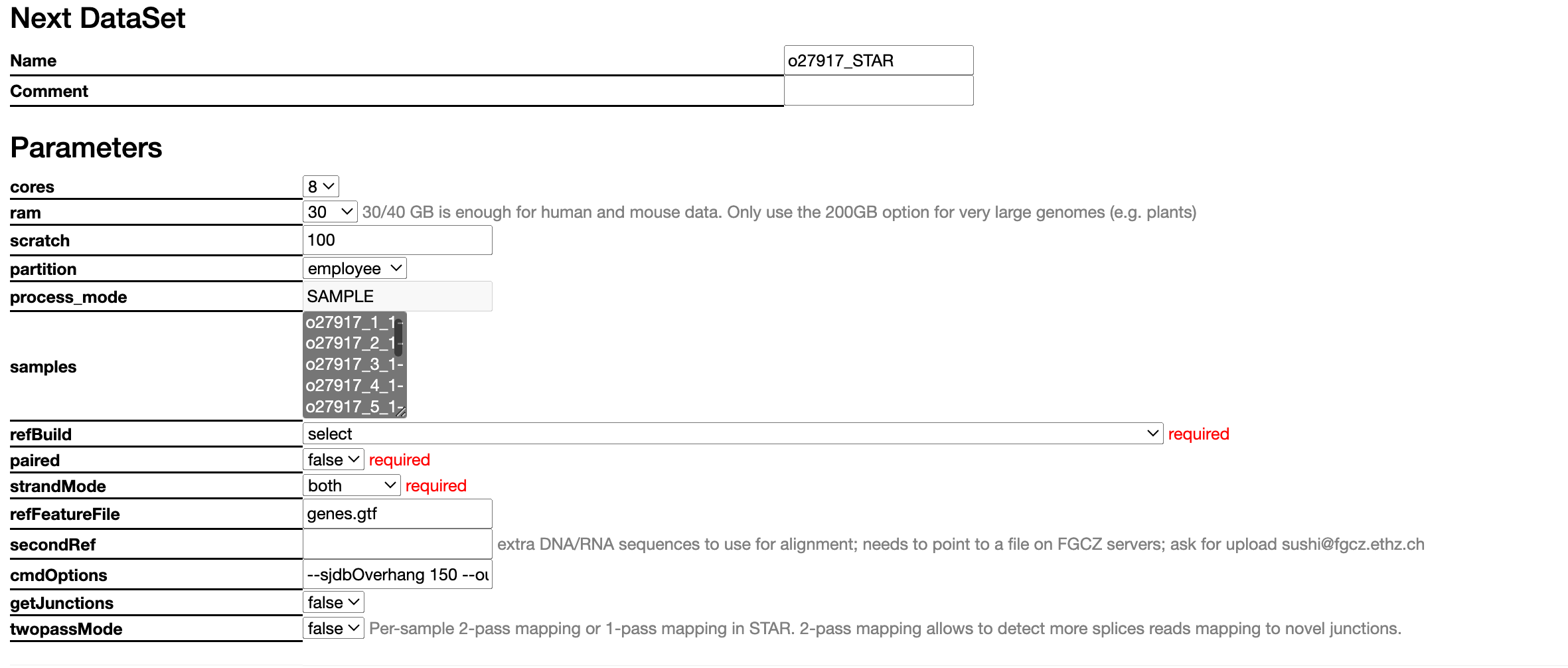
Essential parameters are marked in red. In this case, "paired" and "strandMode" were autopopulated, but "refBuild" was not and will therefore need selecting. Be sure to make good use of the "Comment" box at the top so you can later easily see what parameters you chose.
---
<br><br>
# Static versus Live Reports: What's the difference and which should I use?
- The data that goes into both these reports is identical, as produced by the app you used.
- The static report shows the main results of the analysis when performed, and is not editable/customisable in any way.
- The interactive report shows many of the same results, but allows for customisation of figures, as well as deeper exploration of results.
- The interactive report does not, however, allow for editing of the underlying results. One can make changes to figures, and even exclude certain groups for figures, but not change the results (e.g., p-values).
- We intend users to first closely inspect the static report, before later moving to the interactive report.
---
<br><br>
# Can I accidentally delete all my sequencing data? What happens if I delete something by accident?
- Don't worry! The chances of you irretrievably deleting your raw data are deliberately very low... **but never zero!**
- You can, and should, delete old/broken child DataSets routinely, but they will never delete anything upstream. *E.g.*, if you delete a FeatureCounts DataSet, the parent BAM files remain untouched.
- If you delete a DataSet with children DataSets, *e.g.*, a STAR BAM DataSet that has a child FeatureCounts DataSet, those BAM files will be deleted and the FeatureCounts becomes a new 'root' DataSet, *i.e.*, a top level DataSet.
- But you cannot easily delete a root DataSet, *e.g.*, your raw FASTQ sequencing files. These are the most critical files, and everything else can be generated again.
- If somehow you have managed to delete everything and are beginning to panic, just reach out to us ASAP and we can put it back for you!
---
<br><br>
# Why do my jobs keep failing?
- In most cases, apps fail because of the parameters selected by the user aren't compatible for that DataSet, or something that should have been required snuck through empty.
- Sometimes, the DataSet isn't quite in the right format for the app, *e.g.*, a column is missing.
- But, also, some apps just have bugs in them.
- Please always double check your selected parameters, and reach out to us ASAP for assistance.
---
<br><br>
# How do I know which apps I should use on my data?
- For the most part, we sort of leave it up to you to know what you need to do.
- SUSHI guesses which apps are compatible with your DataSet, but this doesn't mean it's appropriate. Running a metagenome app on your scRNA doesn't seem helpful.
- Try to attend one of our regular SUSHI seminars, day courses, or FGCZ teaching weeks, if you need to learn what steps you need to perform on your data.
---
<br><br>
# What's the easiest way for me to download my raw data?
- The easiest way is to click the "download" link at the top, then click the link next to "Direct http download", and then click to download each file individually. This can be a little arduous if you have very many files, but is the easiest.
- A detailed guide of how to download your data using the FileZilla app can be found here: https://fgcz-intranet.uzh.ch/tiki-index.php?page=bioinformatics.gStore.get_data
- If you want to flex your command line skills, you can copy one of the `wget` or `scp` links also provided in the download box.
- To do this on Mac: Press the command + space keys together, type terminal, and press return (enter) to open the Terminal. Then change the current directory you're in to somewhere useful, like the desktop, by typing in `cd ~/Desktop/` and hitting return (enter). (If you're using a non-English keyboard, you can type `cd /Users/<yourusername>/Desktop` and replace `<yourusername>` with your user name, which you can do by hitting the tab key after /Users/). Then paste in the scp command and hit enter. It will download the whole contents of that DataSet to a folder on your desktop.
- To do this on Windows: Press the start key and type "Powershell" and open it. Then type in `cd C:\Users\<YourUsername>\Desktop`, and replace `<YourUsername>` with your user name, which you can do by hitting the tab key after `\Users\`. Then paste in the scp command and hit enter. It will download the whole contents of that DataSet to a folder on your desktop.
- If you're still having troubles using the command line to download your files, let us know!
---
<br><br>